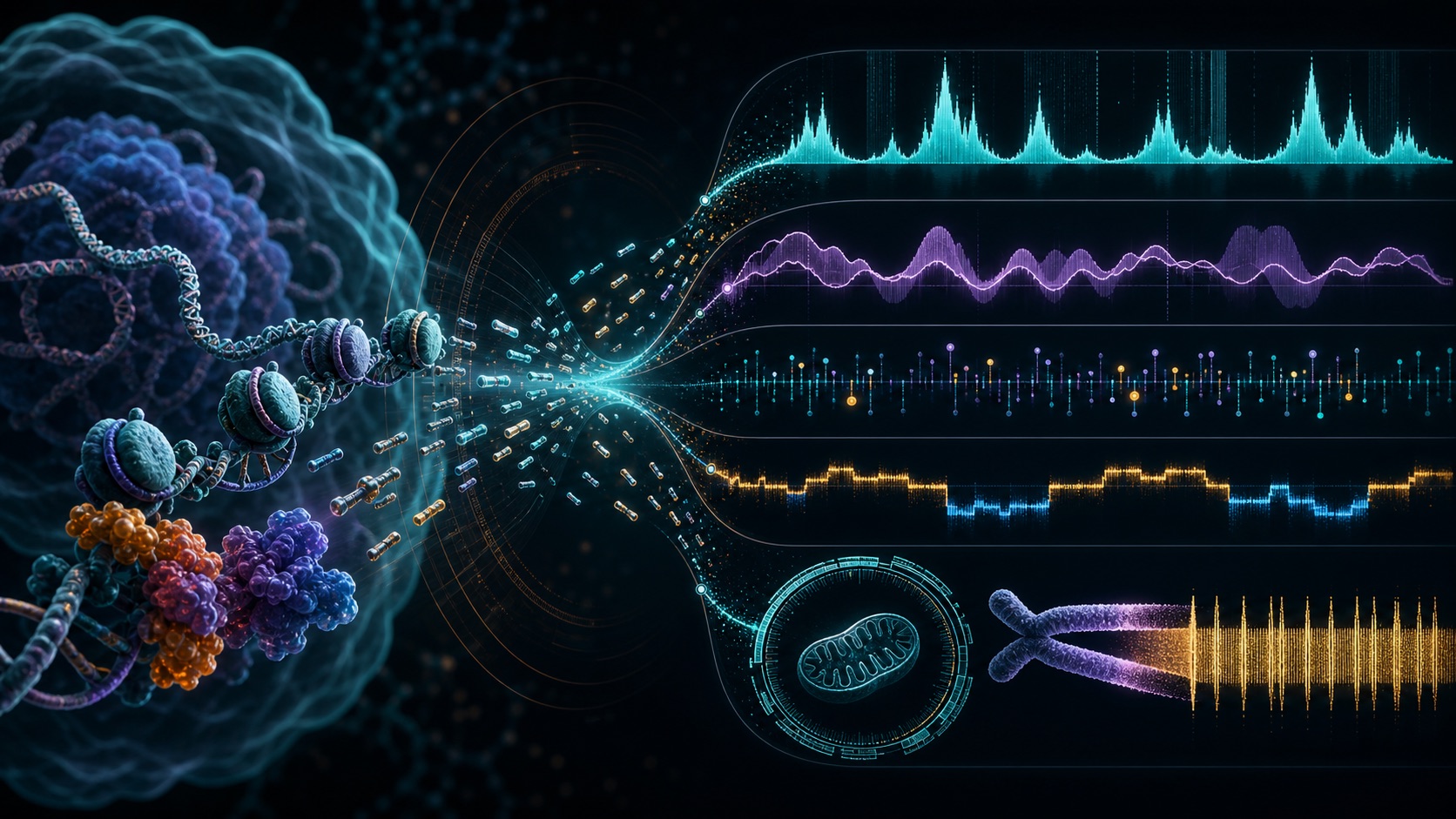
ATAC-seq is widely used to map chromatin accessibility, but bulk ATAC-seq libraries contain much more information than accessible regulatory elements alone. In recent work, I benchmarked ATAC-seq against matched whole-genome sequencing to evaluate whether a single assay can also recover genetic information relevant to cancer and aging biology.
Using paired datasets from patient-derived melanoma cell lines and TCGA brain tumors, we showed that ATAC-seq can support high-precision small variant detection within accessible regions, robust copy-number profiling, mitochondrial genome characterization and telomere-associated repeat quantification, while preserving its classical epigenomic readouts. This establishes ATAC-seq as a cost-effective dual-purpose platform linking genome integrity, chromatin state and regulatory network activity.
Building on this concept, I am developing the eGATTACA framework: epigenetic and genetic aging and tumor tracking through ATAC-seq for clinical assessment. The goal is to systematically extract, harmonize and model genetic and epigenetic features from ATAC-seq profiles to build interpretable signatures of aging trajectories, frailty risk and tumor evolution.
The project is designed to move from mechanistic models to clinically grounded cohorts. In aging mouse models, ATAC-seq-derived features will be integrated with metabolic responses and p53-dependent regulatory programs. In human primary fibroblasts from the INSPIRE cohort, the same framework will be used to connect chromatin accessibility, genomic instability, mitochondrial features and telomere-associated signals with metabolic phenotypes and clinical frailty indices.