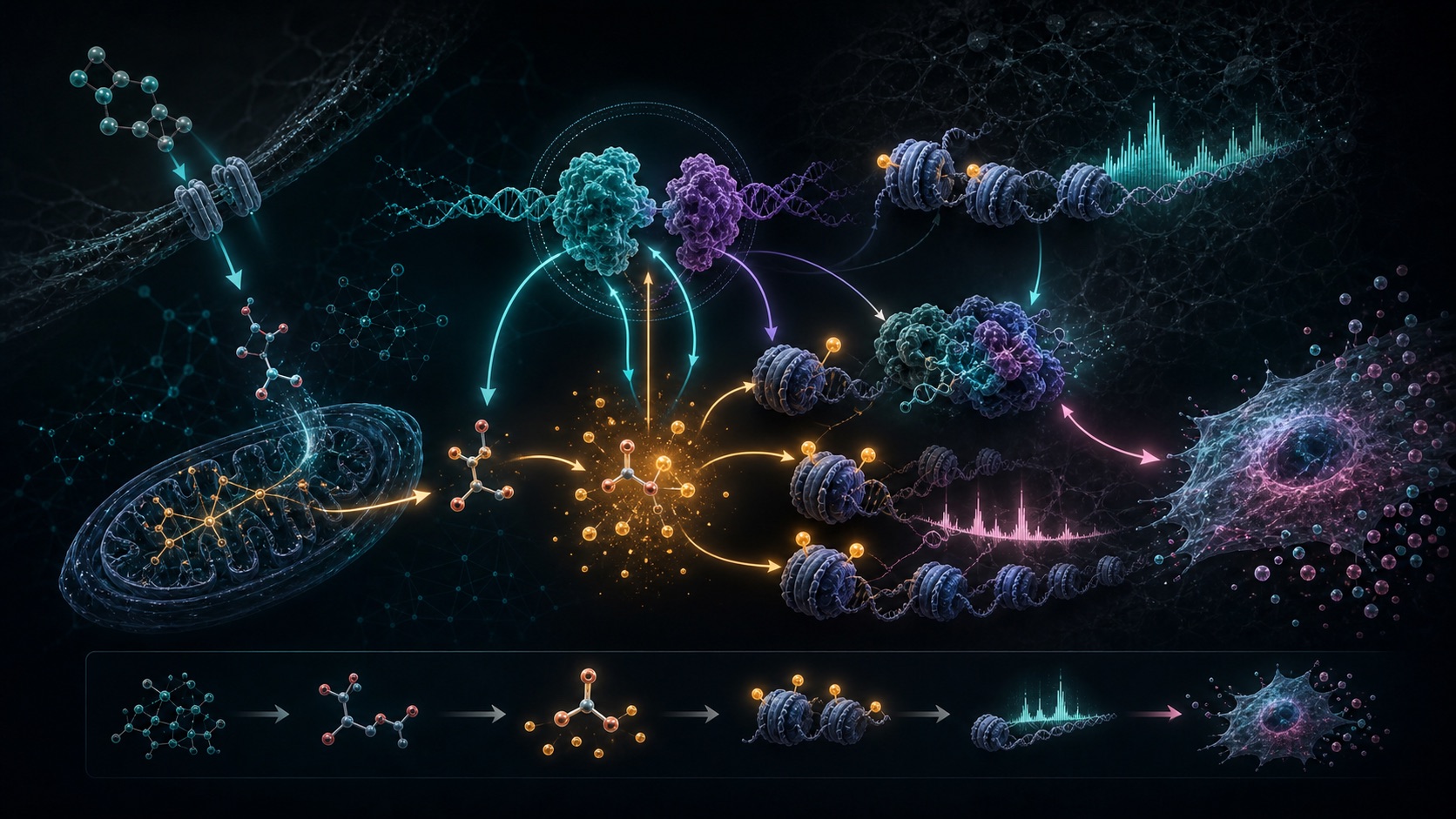


A practical way to prioritize transcription factors by combining motif enrichment, expression, chromatin accessibility, timing and network position.
Metabolism is not merely a source of energy: it continuously instructs gene regulation by shaping chromatin marks and transcriptional programs. Over the years, I have progressively investigated this metabolism-gene regulation axis across complementary biological scales and questions - from skin physiology and microbiome-host interactions to the regulation of cellular senescence and its pathophysiological consequences in aging and cancer. Along this path, I have pursued integrative studies bridging genetics, functional genomics, and systems modeling to understand how metabolic rewiring reshapes regulatory networks in diverse physiological and pathophysiological settings. These projects have provided me with strong expertise in molecular and cellular biology as well as computational biology, including bioinformatics, data integration, and network inference. At the interface of experimental biology and computation, I have developed an integrated view of how metabolic states translate into epigenetic and transcriptional programs that govern cell fate decisions, including senescence. Moving forward, I will build on this framework to address pathophysiological questions at the intersection of metabolism, epigenetics, and aging.
I am currently working at the Montpellier Cancer Research Institute as a postdoctoral fellow, in the unit Molecular Oncogenesis headed by Laurent Le Cam.
Interests
- Functional genomics
- Genetics
- Epigenomics
- Metabolism
- Systems biology
- Cellular senescence
- Cancer biology
Education
-
PhD in Genetics and Genomics, 2014
Agrocampus Ouest, Rennes, France
-
MEng in Agronomical Science, 2011
Agrocampus Ouest, Rennes, France
-
MSc in Cell and Molecular Biology, 2011
Université Rennes 1, Rennes, France
-
MSc in Environment and Oceanology, 2009
Université des Sciences et Technologies, Lille, France
Hobbies
- Photography
- Drawing
- Scuba diving
- Botany
- Traveling
CNU
- CNU 64: biochemistry and molecular biology
- CNU 65: cell biology